Last updated: 2025-07-31
Checks: 7 0
Knit directory: GradPLog/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220610) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 01ec7ef. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/rotation.Rmd) and HTML
(docs/rotation.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 01ec7ef | “John | 2025-07-31 | update progression |
| html | f714464 | “John | 2025-07-31 | Build site. |
| Rmd | a2aa03f | “John | 2025-07-31 | update progression |
| html | 1be3419 | “John | 2025-06-25 | Build site. |
| Rmd | 832fd41 | “John | 2025-06-25 | update rotation project |
| html | caf3504 | zq2209 | 2025-06-25 | Build site. |
| Rmd | 615a021 | zq2209 | 2025-06-25 | update rotation page |
| html | 642ac0b | zq2209 | 2025-06-25 | Build site. |
| Rmd | a0a1f62 | zq2209 | 2025-06-25 | update rotation page |
| html | a0a1f62 | zq2209 | 2025-06-25 | update rotation page |
| Rmd | ae3d616 | zq2209 | 2025-06-25 | update rotation page |
| html | ae3d616 | zq2209 | 2025-06-25 | update rotation page |
| Rmd | 1d71192 | zq2209 | 2025-06-25 | update rotation page |
| Rmd | bd405e1 | zq2209 | 2025-06-25 | update rotation page |
| html | bd405e1 | zq2209 | 2025-06-25 | update rotation page |
| Rmd | 509f6ca | zq2209 | 2025-06-25 | update rotation page |
| html | 509f6ca | zq2209 | 2025-06-25 | update rotation page |
| Rmd | 783dfdb | zq2209 | 2025-06-25 | add ppi rotation project |
Goal
Investigate potential mechanisms that may underlie genetic variants that affect a gene’s protein expression level in trans, but not its mRNA expression level.
Curate databases that capture different mechanisms including annotated strict protein complex partners, many different methods that identify protein-protein interaction networks, known signaling pathway, and more.
Develop approaches that may help distinguish trans-pQTLs that are driven by cell-composition effects versus intra-cellular trans-pQTL effects
Jun 26 Update
Test whether pQTL enriched in protein pairs
Dataset
PPI
Bioplex: ~ 2900 pairs after overlapping with pQTL data
Hippie: ~ 1346 pairs after overlapping
Corum: ~ 1270 pairs after overlapping
String: ~ 3391 pairs after overlapping
Gene module
DNG gene module by trans-PCO:
166 gene modules
12000+ genes in total
over 1,000,000 gene pairs
QTL summary statistics
UKB-PPP: plasma
DGN: whole blood
Prodecure
For each gene/protein pair, pick up the SNP from the most significant transQTL (i.e. smallest pvalue) of gene/protein A.
For gene/protein B of this gene pair, pick up pvalue of QTL for this SNP and B.
After fisrt 2 steps, there will be a pair of pvalues, then calculate the proportion that both pvalues are significant under certain pvalue threshold, i.e. the probability of there is transQTL for B given there is transQTL for A.
Use bootstrap to somehow mimic the distribution of the probability. For each dataset, each time select certain number of pairs and calculate probability of this dataset. From boostrap, there will be distribution of probability, able to estimate standard errors and construct confidence intervals.
Note: Not sure about which pvalue threshold and sample size of bootstrap, so have results for multiple pvalue thresholds and boostrap sample sizes.
Preliminary result
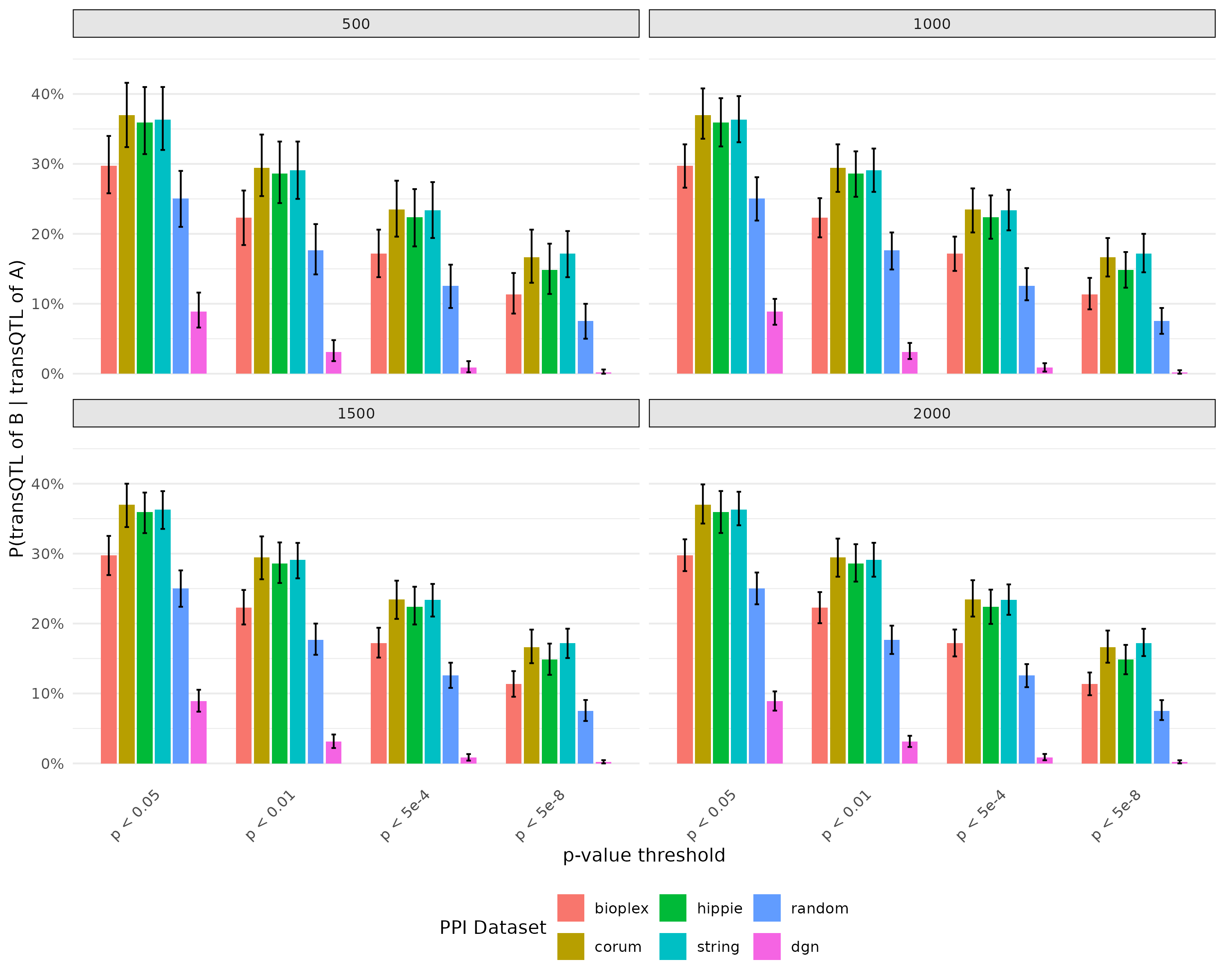
- Plot for probability of there is transQTL for B given there is transQTL for A with bootstrap
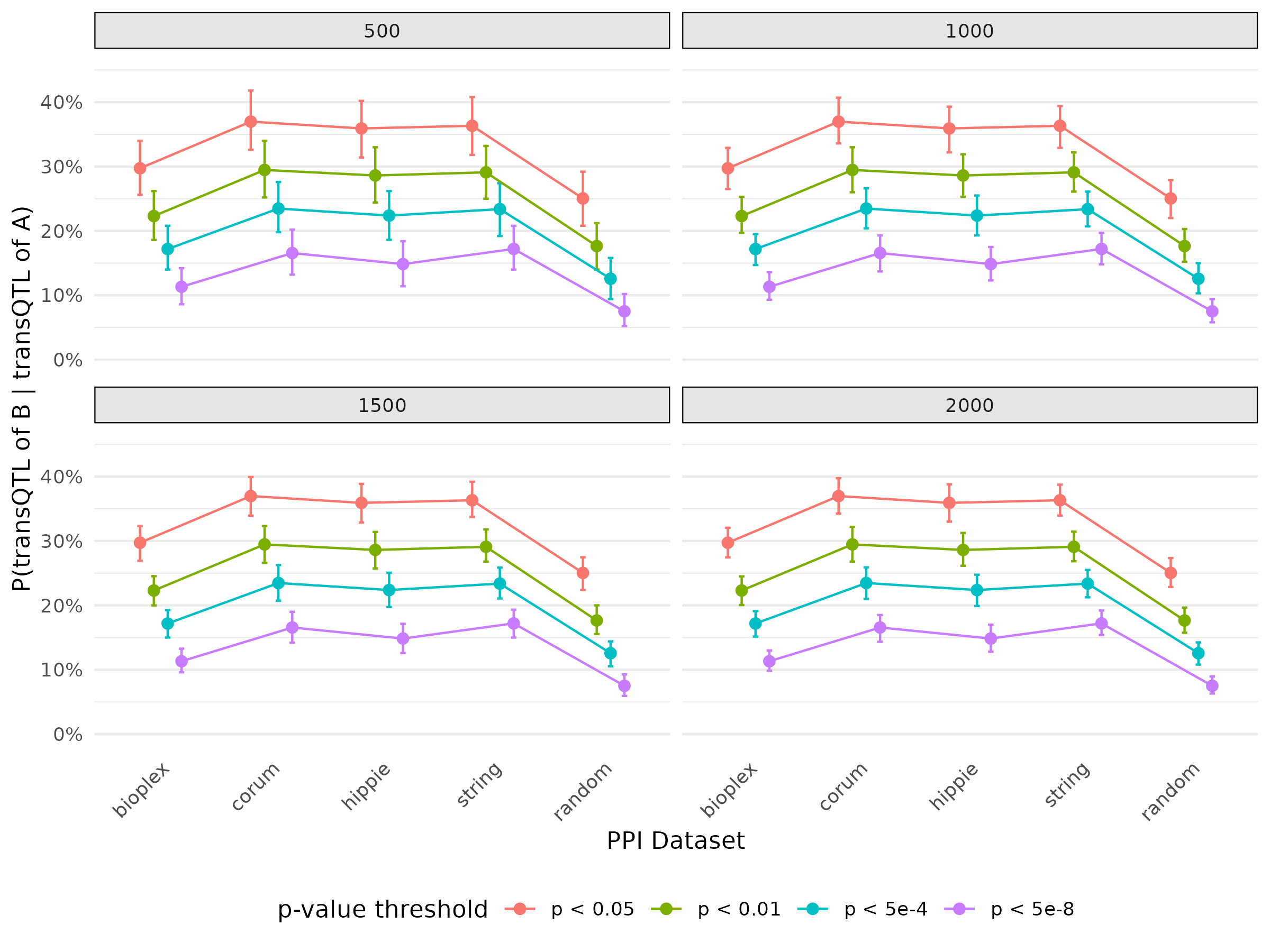
The overall trend as expected, PPI pairs have higher probability then random pairs. And the error bar decrease as sample size of bootstrap increase.
- Table for cisQTL for B given there is transQTL for A
| Dataset | Total | trans.cis |
|---|---|---|
| bioplex | 5800 | 21 |
| corum | 2542 | 25 |
| hippie | 2692 | 11 |
| string | 6698 | 63 |
| random | 4471 | 6 |
Note: Total pairs is the number of unique pairs * 2
Consideration & Disscussion
- The tissue difference may have impact on the result
Different tissue of PPI datasets:, e.g whole blood cells for bioplex and general cells for others
Different tissue in pQTL and eQTL data
- The technology difference may also have some impact
- e.g. bioplex test for interaction instead of complex
- The difference in number of pairs
July 31 - Update
- Plot for probability of there is transQTL for B given there is transQTL for A with bootstrap - Updated
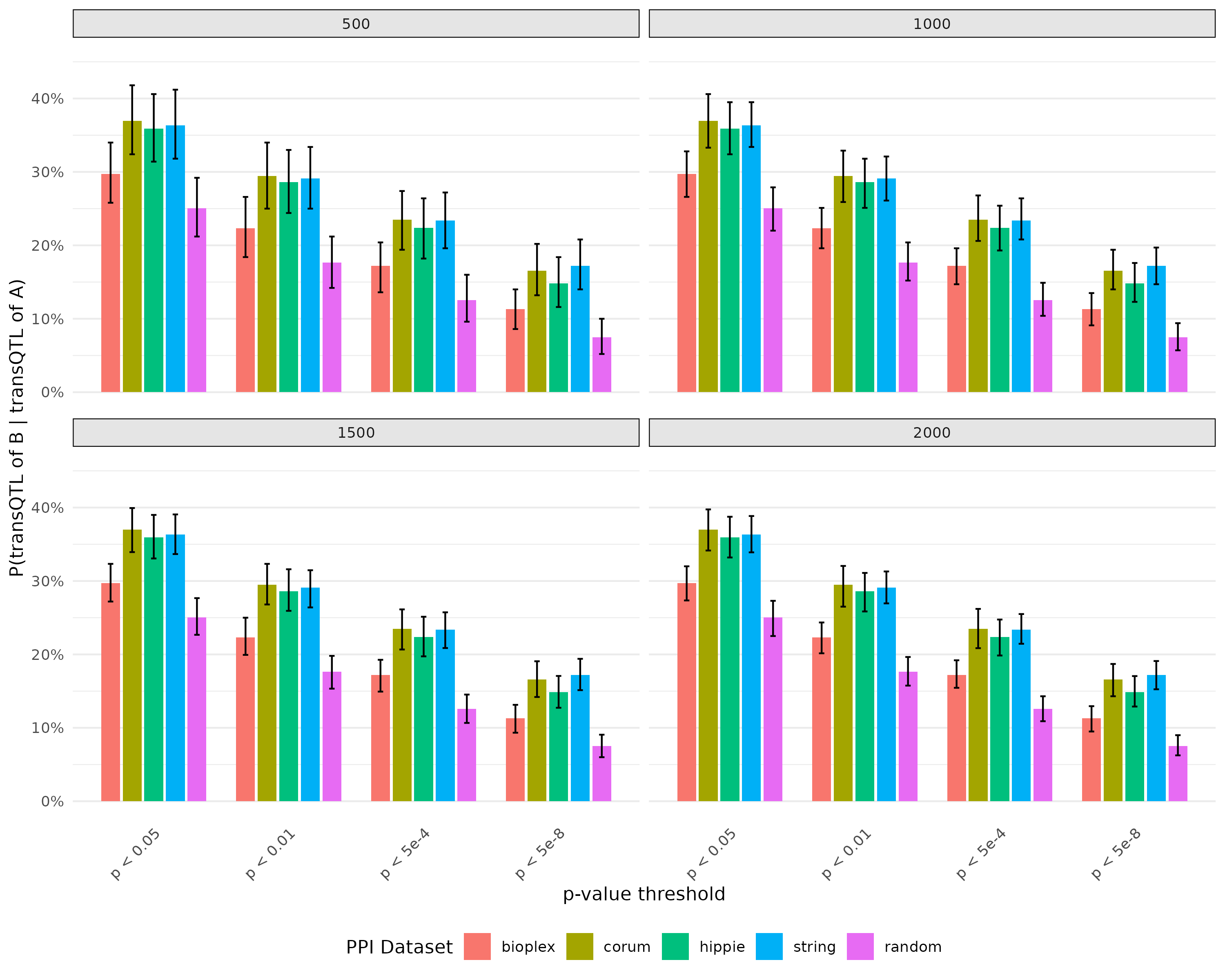
Exclude SNPs colocalized with cell type composition
Plot for probability of there is transQTL for B given there is transQTL for A with bootstrap
- Exclude SNPs colocalized with cell type composition
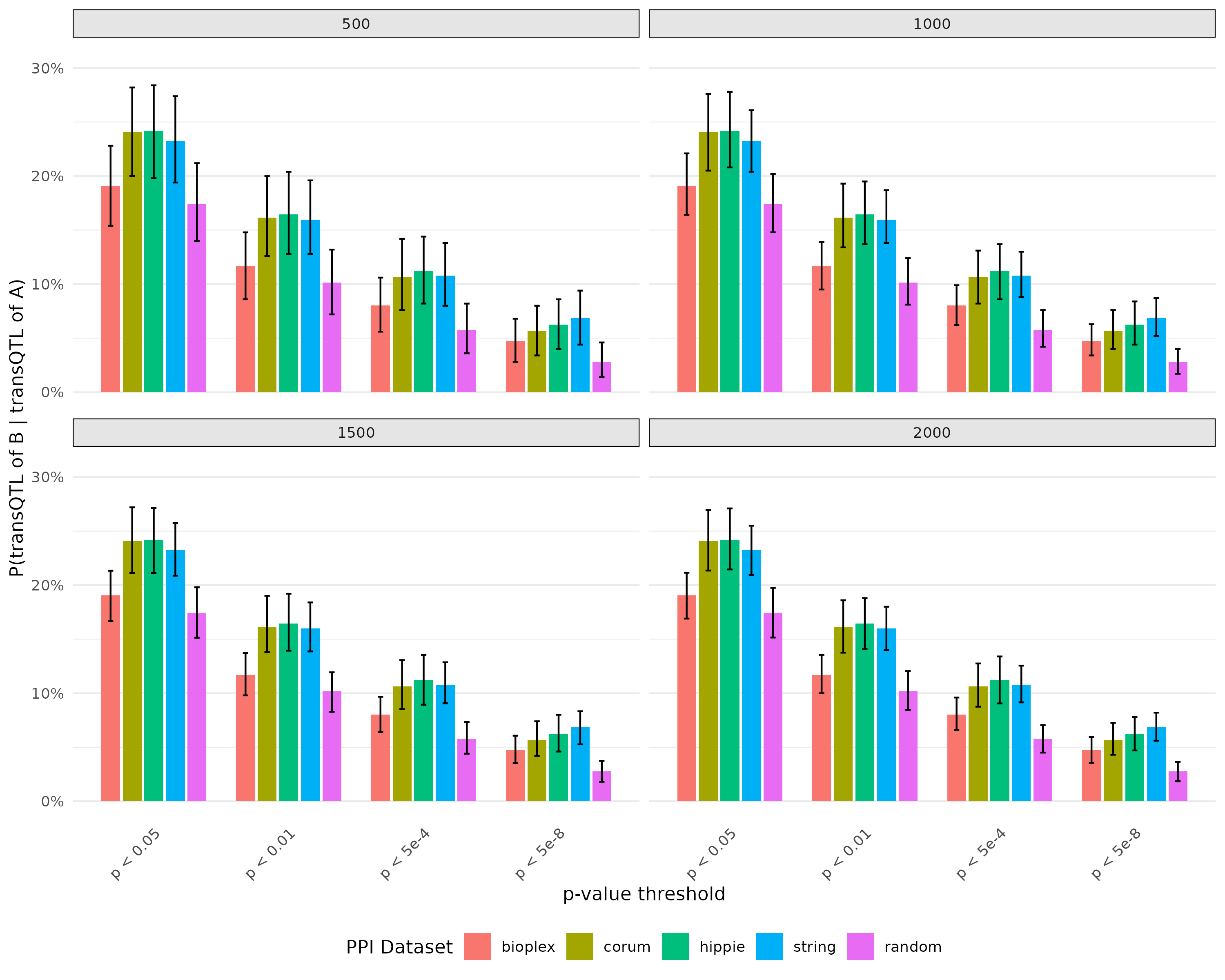
The average proportion are relatively lower than before. But the trend remain the same.
Test how many unsignificant snp for protein A is significant for protein B
Plot for probability of there is transQTL for B given there is non-transQTL for A with bootstrap
Same procedure as before but first extract snp that not significant for protein A
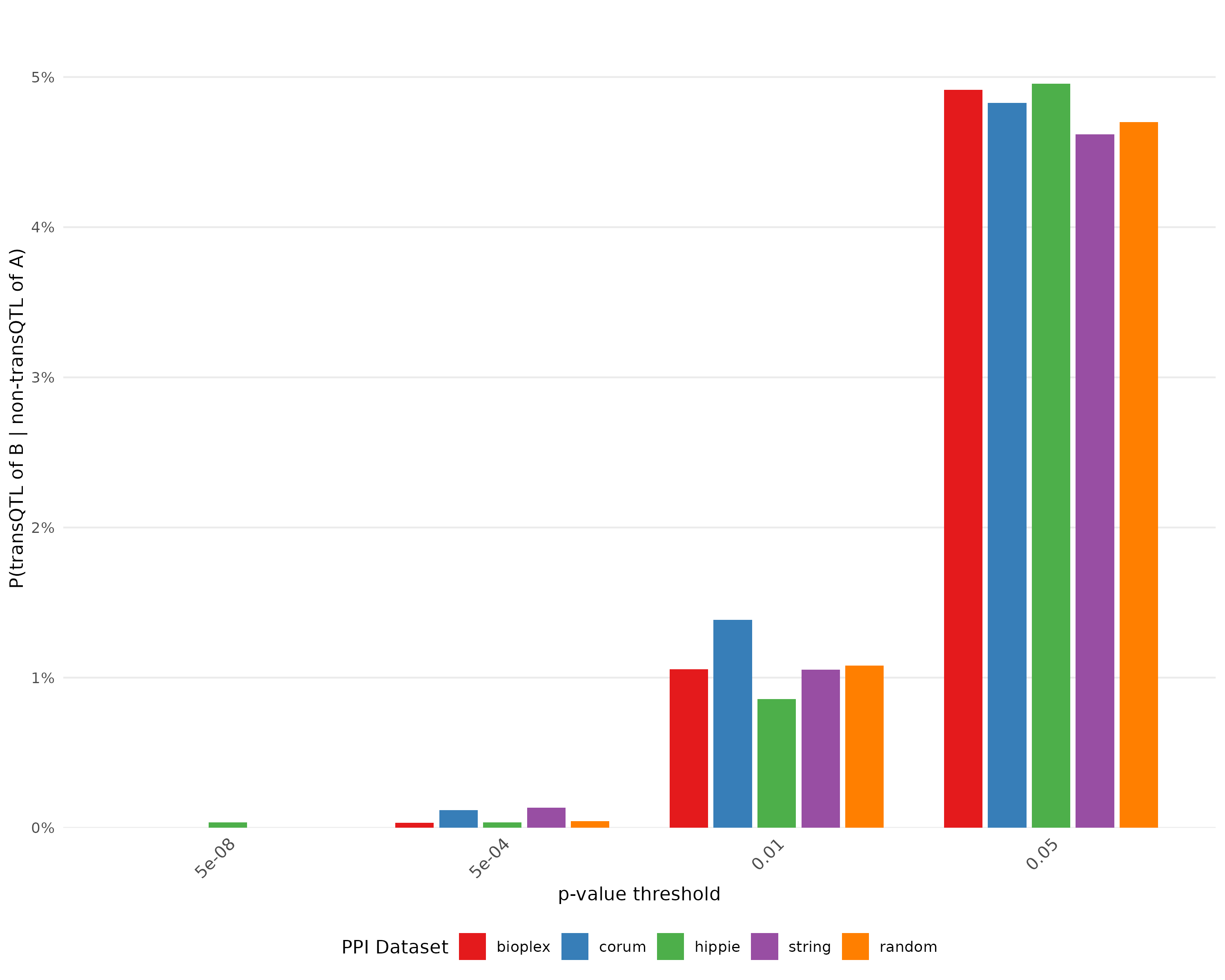
From the plot, the proportion of transQTL for B given non-transQTL for A is close to corresponding threshold.
Analysis on top 2 significant snps
This is to check whether most significant SNP for A is less than the cutoff when second significant SNP for A is less than the cutoff
For each pair, choose the top 2 significant SNP for A
check whether the SNP 2 (second significant snp) is less than cutoff for B
Check whether SNP 1 is less than cutoff when SNP 2 is less than cutoff or not
| snp1 significant | snp1 not significant | Total | |
|---|---|---|---|
| snp2 significant | 1507 (0.89) | 192 (0.11) | 1699 |
| snp2 not significant | 225 (0.05) | 3874 (0.95) | 4099 |
| snp1 significant | snp1 not significant | Total | |
|---|---|---|---|
| snp2 significant | 813 (0.88) | 116 (0.12) | 929 |
| snp2 not significant | 126 (0.08) | 1480 (0.92) | 1606 |
| snp1 significant | snp1 not significant | Total | |
|---|---|---|---|
| snp2 significant | 853 (0.91) | 81 (0.09) | 934 |
| snp2 not significant | 118 (0.07) | 1640 (0.93) | 1758 |
| snp1 significant | snp1 not significant | Total | |
|---|---|---|---|
| snp2 significant | 2160 (0.91) | 217 (0.09) | 2377 |
| snp2 not significant | 324 (0.07) | 4060 (0.93) | 4384 |
| snp1 significant | snp1 not significant | Total | |
|---|---|---|---|
| snp2 significant | 907 (0.83) | 185 (0.17) | 1092 |
| snp2 not significant | 212 (0.06) | 3165 (0.94) | 3377 |
Based on the result, simple chi-square test is performed. Chi-square tests are significant, which means that whether SNP1 is significant is associated with whether SNP2 is significant or not.
Analysis on gene pairs
Plot for probability of there is transQTL for B given there is transQTL for A with bootstrap
Same procedure as before
Next step
Same thing in eQTL and CSF pQTL
Any other PPI module
- Production of Protein-Complex Components Is Stoichiometric and Lacks General Feedback Regulation in Eukaryotes: Protein complex from four species.
R version 4.4.3 (2025-02-28)
Platform: aarch64-apple-darwin20
Running under: macOS Sequoia 15.4.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] dplyr_1.1.4 kableExtra_1.4.0 knitr_1.50
loaded via a namespace (and not attached):
[1] jsonlite_2.0.0 compiler_4.4.3 promises_1.3.2 tidyselect_1.2.1
[5] Rcpp_1.0.14 xml2_1.3.8 stringr_1.5.1 git2r_0.36.2
[9] later_1.4.2 jquerylib_0.1.4 textshaping_1.0.1 systemfonts_1.2.3
[13] scales_1.4.0 yaml_2.3.10 fastmap_1.2.0 R6_2.6.1
[17] generics_0.1.4 workflowr_1.7.1 tibble_3.2.1 rprojroot_2.0.4
[21] svglite_2.2.1 bslib_0.9.0 pillar_1.10.2 RColorBrewer_1.1-3
[25] rlang_1.1.6 cachem_1.1.0 stringi_1.8.7 httpuv_1.6.16
[29] xfun_0.52 fs_1.6.6 sass_0.4.10 viridisLite_0.4.2
[33] cli_3.6.5 magrittr_2.0.3 digest_0.6.37 rstudioapi_0.17.1
[37] lifecycle_1.0.4 vctrs_0.6.5 evaluate_1.0.3 glue_1.8.0
[41] farver_2.1.2 whisker_0.4.1 rmarkdown_2.29 tools_4.4.3
[45] pkgconfig_2.0.3 htmltools_0.5.8.1